Molecular Engineering of Tools for in vivo Genome Manipulation

Figure 1: E. coli expressing a fluorescently labeled protein. Here we're expressing a tagged protein that binds to mRNAs so that we can watch the RNAs diffusing around!
Request plasmids for use in your lab from Addgene here!
One of the basic procedures of experimental biology is to introduce genes into your organism of interest and study the effects of their expression. For example, you might want to tag a protein with green fluorescent protein (GFP) and express it in E. coli; if you could put this tagged protein into the cell and look at it under the microscope, you could measure how much of the protein is produced by looking at the total fluorescence in the cell, or you could measure protein localization by looking at what areas in the cell were more fluorescent than others. But first you've got to get the gene encoding the tagged protein into the cell so that E. coli can make it for you!
An easy way to do this is to put your gene onto a plasmid. It is extremely easy to get plasmids into E. coli, but there are disadvantages: generally, once you get the plasmid into the cell, its replication and maintenance means that the cells have to do extra work to keep the plasmid, which they don't like to do. That means you've got to force them to keep it, and this is usually done by also including a gene on the plasmid that confers resistance to some antibiotic. Then, by growing your bacteria in medium that includes this antibiotic, only those cells that maintain the plasmid and its accompanying antibiotic resistance gene can survive and grow. However, the antibiotic and the energetic load required to express the protein making them resistant can still influence the growth of the cell, which is not ideal for careful experiments. Also, plasmids generally exist in the cell in 1-1,000 copies, depending on the type of plasmid you're using. But these are average copy numbers; due to stochastic fluctuations, individual cells may have significantly more or less plasmid than this average number, and these random fluctuations can make life difficult when trying to interpret your data.
Therefore, it can be extremely advantageous to put your gene directly into the organism's chromosome to eliminate these difficulties. Once your gene is in the chromosome it is stable, and there's no need to use antibiotic selection to force the cells to keep it. Moreover, the replication of the chromosome is highly controlled and well understood, so that you don't have to worry (too much) about copy number fluctuations.
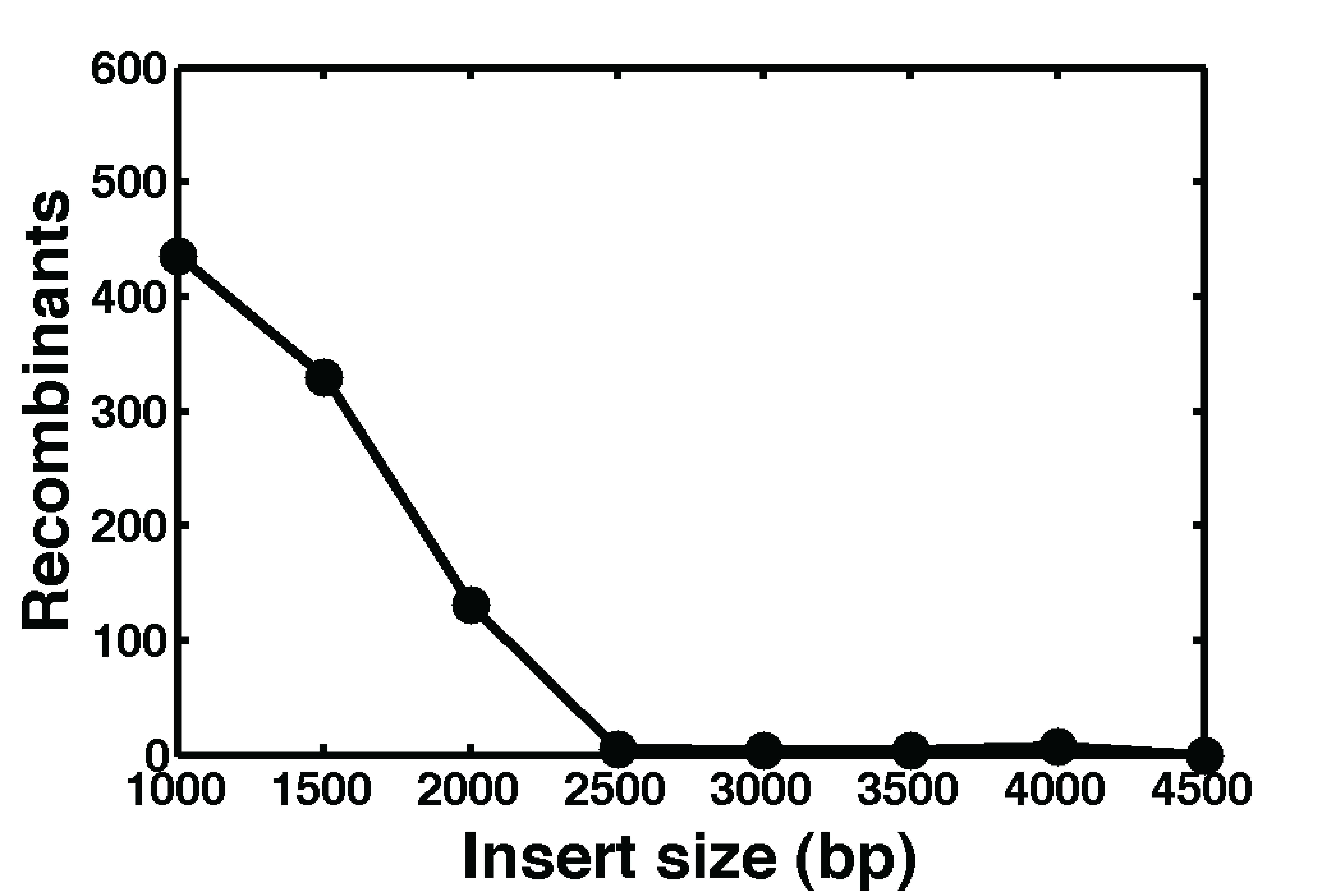
Figure 2: An example of the efficiency of recombineering as a function of insert size. For things larger than ~2500 base pairs, it becomes progressively more difficult to obtain successful integrants.
However, getting genes into the chromosome is not as easy as giving the bacteria a plasmid! There are two primary technologies people currently use to do this: recombineering, and phage-derived methods. But each of these procedures has drawbacks: recombineering is extremely easy and effective, and you are able to designate exactly where in the chromosome your gene will go. However, recombineering generally only works reliably for small genes (< ~2500 base pairs). Phage-derived methods have the opposite problem: you can put things in that are as big as you want, but without a lot of work you are limited to incorporating it into only a few pre-existing integration locations.
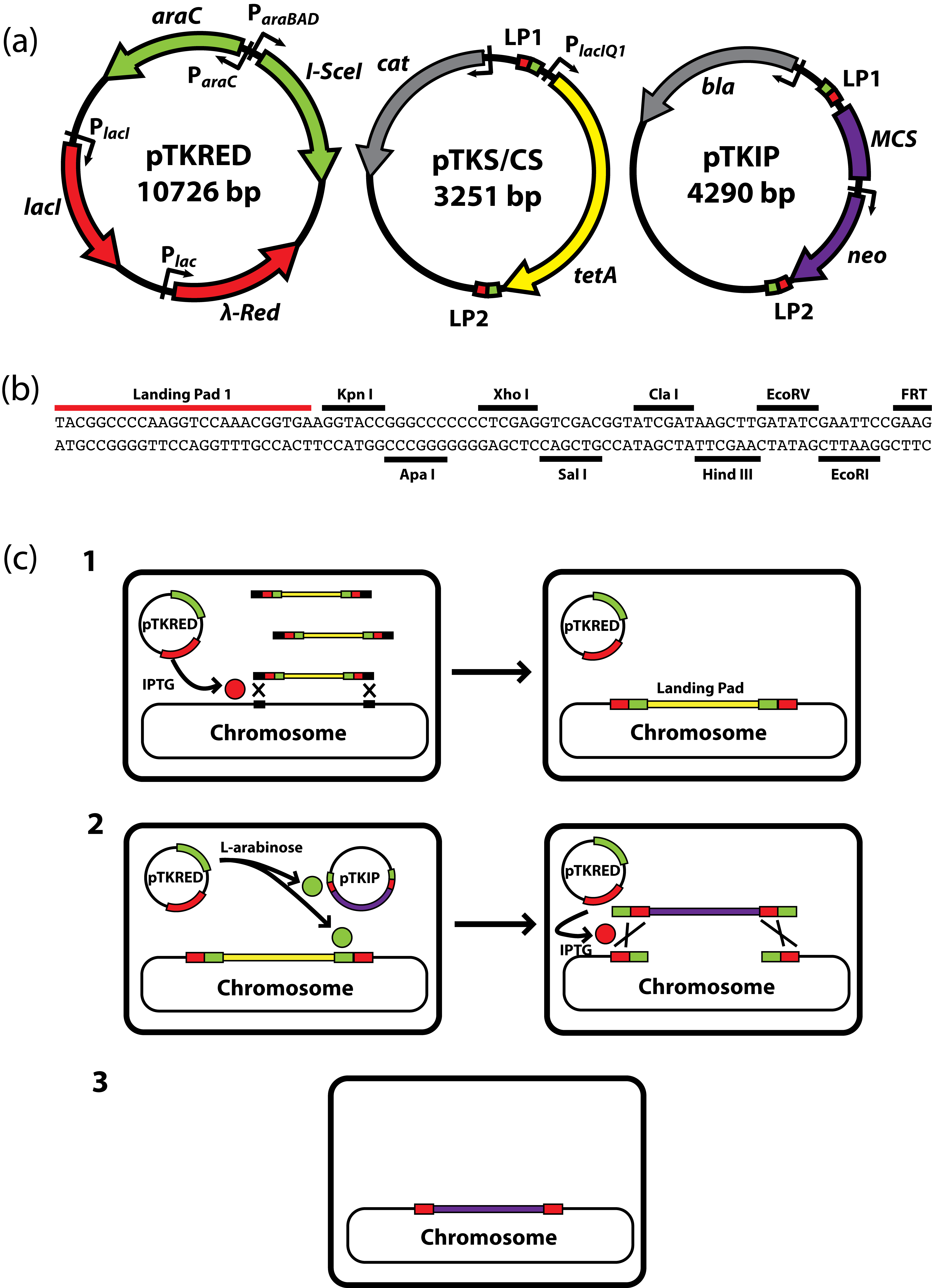
Figure 3: Plasmids and procedure for Landing Pad-based chromosomal integration.
We have developed a simple and easy two-step method that combines recombineering with in vivo sub-cloning for integrating genes into the E. coli chromosome. This procedure combines the advantages of both recombineering and phage-based methods with none of their drawbacks: we are able to precisely integrate very large fragments of DNA into any desired location and orientation in the chromosome. The first step is to integrate a small (~1300 base pair) "landing pad" into the desired integration location in the chromosome by recombineering. You then transform the cells with an engineered plasmid that contains your large integration fragment. Then, with a bit of simple biological wizardry, your large integration fragment is excised from the plasmid in vivo and integrates into the chromosome at the landing pad.
This system works very easily and effectively in E. coli, and we have distributed the plasmids to dozens of labs around the world and licensed them to several biotech companies. But there is room for improvement, and we are currently working on upgrades of the system and expanding its applicability to additional organisms.